TUTORIAL
Welcome to tutorial page of MaSH~
Here you can begin with your precious samples and gain and understanding of mass spectrometry analysis options at the University of Auckland.
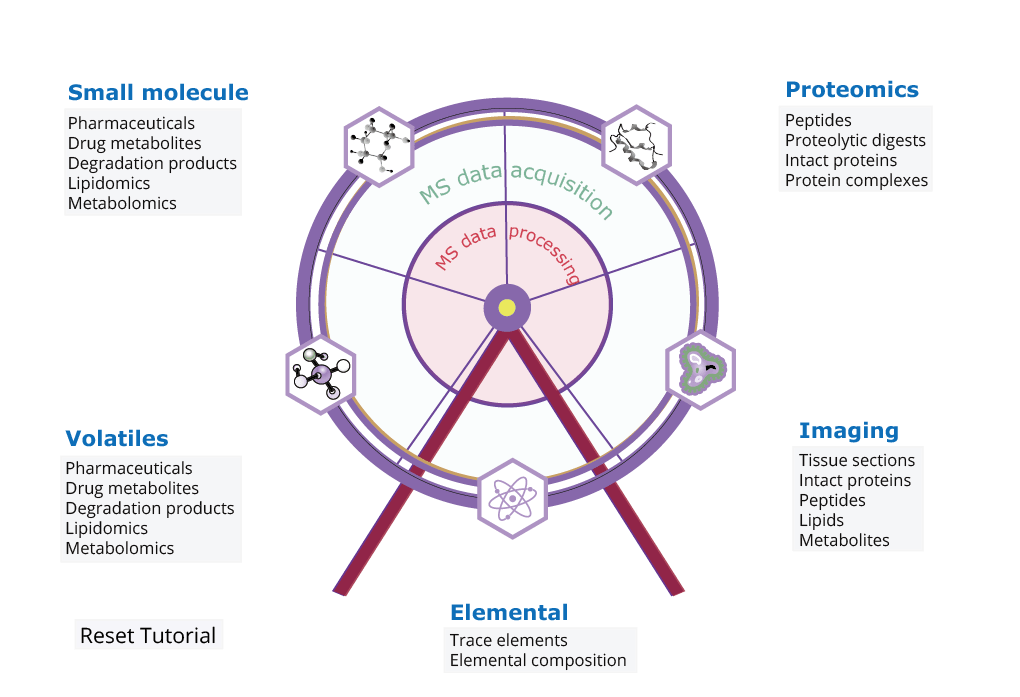
Step1: Click on your sample type and check out the general requirements for MS analysis of different (bio)molecules. This will give you a brief overview of sample preparation and sample analysis conditions.
Step2: Next, click on the outer wheel to check out instrument options and get in touch with the right people.
Step3: Finally, click the inner wheel region to find the appropriate data processing pipelines that help you out. We provide both open source and commercially available data solutions for you. Alternatively, you are able to build your own one through the OpenMS KNIME infrastructure.
Have fun!
*This is a general tutorial and may not include your specific application – if you can’t find what you’re looking for, don’t hesitate to Get in touch with MaSH to see if there are still options for you.
Proteomics MS

For proteomics profiling, SWATH and PRM analysis: LC-Triple-TOF 6600
For general peptide analysis, method development: LC-LTQ Ion Trap and LC-QSTAR XL
Contact person: Martin Middleditch
For quantitative proteomics analysis :
LC-Triple-Quadrupole Agilent 6460
Contact person: Githal Arachchige
LC-Triple-Quadrupole Agilent 6410
Contact person: Sree Sreebhavan and Frederik Pruijn
Small molecule MS

LC coupled with Quadropole-Orbitrap MS: Quadropole MS analyzer gives you good quantitative reproducibility. Orbitrap holds a high m/z resolution with fast scan speed. This system suits most of the small molecule cases.
Contact person: Kalita Prangnell
Contact person: Eric Thorstensen and Kalita Prangnell
If your sample doesn't need a LC separation, MALDI MS will give you quick and promising result at reasonable price:
Contact person: Githal Arachchige
Proteomics data processing
For the metabolically labelled sample, you can use Quantitative proteomics pipeline to analysis your data. You can also use the label-free or SWATH workflow to get a semi-quantitative result. You need to prepare a suitable database based on your proteomics target including any putative proteins that could exist in your sample. That will be a huge help to reduce the false discovery rate.
Choose appropriate workflow and modify it to fit your experimental design and statistical requirement
1. Label-free protein quantification: Workflow
2. Targeted proteomics analysis: Workflow
3. Quantitative proteomics analysis: Workflow
Click for more details and software on our virtual machine: virtual machine list
Peptides
Sample Clean-up: Filter sample prior to sample submission
Loading amount: 1~5 ug
Mass range: 1000Da to 6000Da
Chromatography system: Column and running buffer
MS Resolution:
Use low to middle-resolution MS analyzer for quantitative analysis. Use middle to high resolution for qualitative analysis.
Low res: Triple-quadrupole MS, LTQ MS
Middle res: Triple-TOF, Q-TOF
High res: Orbit-trap, FT-ICR
Intact protein
Intact protein has the mW with over 20000 Da.
There are commercially available columns for direct protein analysis.
Use low-resolution mode(~10000) if you want to get glycosylation profile.
Use ultra resolution mode if you want to get the accurate mW.
Volatile MS

If you are analyzing volatile molecules, use the gas chromatography based MS to resolve the sample. Make sure that the sample has been derivatized and pre-cleaned. GC linked with quadrupole analyzer will give you promising analysis performance for molecules under 1000 Da.
Contact person: Jin Wang and Saras Green
Contact person: Githal Arachchige
Elemental MS

Inductively Coupled Plasma Mass Spectrometry (ICP-MS) is a technique suitable for trace elemental analysis of several materials, including geological, biological, metallurgic, agricultural and environmental samples. Its main advantage is that it can analyse a large number of elements in a single analytical run. Samples can be analysed in either a liquid (solution ICP-MS) or solid form (laser ablation; LA-ICP-MS) with typical detection limits for most elements in the parts per trillion range.
Click for more details: Inductively Coupled Plasma Mass Spectrometry
Contact person: Stuart Morrow
Imaging MS

-Capable of imaging all types of biomolecules (e.g. proteins, peptides, lipids, metabolites, drugs)
-User training available
Contact person: Gus Grey and Richard Yulo
Imaging MS data processing
You could use DataAnalysis; BioTools and Sequence Editor for proteomics orientated MALDI data analysis.
FlexImaging and SCiLS Lab are specially designed to address the MALDI imaging data analysis task.
We have developed in-house scripts to automate spatial quantitative and qualitative data processing.
Contact George GUO for more information
Click for more details and software on our virtual machine: virtual machine list
sc-cerndem01.uoa.auckland.ac.nz
mdscils01.uoa.auckland.ac.nz
mdscils02.uoa.auckland.ac.nz
mdscils03.uoa.auckland.ac.nz
Elemental MS data processing
Agilent Mass Hunter controls the ICP-MS. All data output is in an excel chart.
Contact person: Stuart Morrow
Click for more details and software on our virtual machine: virtual machine list
sc-cer00254.uoa.auckland.ac.nz
Volatiles MS data processing
Spectral library: Wiley Nist 2017 Mass Spectral Database
Software: MassHunter, Chemstation, AMDIS, NISTMS, R, Python, ChemOffice
Pipeline: MassOmics, MS-DIAL, MzMine 2
We have developed an in-house pipeline "MassOmics" to automate the untargeted metabolomics data processing.
Contact George GUO for untargeted metabolomics data analysis.
Contact Jin Wang and Saras Green for metabolomics or targeted volatiles data analysis.
Click for more details and software on our virtual machine: virtual machine list
metabolomics.uoa.auckland.ac.nz
Small molecule MS data processing
Spectral library: Wiley NIST08/ NIST17 Mass Spectral Database
Software: MassHunter, Chemstation, AMDIS, NISTMS, ChemOffice, SIEVE, Xcalibur
Pipeline: Lipid search, MS-DIAL with lipid blast
Contact George GUO and Kalita Prangnell for metabolomics and lipidomics analysis.
Contact Jin Wang and Kalita Prangnell for LC-based small molecule analysis.
Click for more details and software on our virtual machine: virtual machine list
qexdp.cer.auckland.ac.nz
sc-cer00254.uoa.auckland.ac.nz
TUTORIAL
Welcome to the tutorial page of MaSH~
Here you can begin with your precious samples and gain an understanding of mass spectrometry analysis options at the University of Auckland.
Step1: Click on your sample type and check out the general requirements for MS analysis of different (bio)molecules. This will give you a brief overview of sample preparation and sample analysis conditions.
Step2: Next, click on the outer wheel to check out instrument options and get in touch with the right people.
Step3: Finally, click the inner wheel region to find the appropriate data processing pipelines that help you out. We provide both open source and commercially available data solutions for you. Alternatively, you are able to build your own one through the OpenMS KNIME infrastructure.
Have fun!
*This is a general tutorial and may not include your specific application – if you can’t find what you’re looking for, don’t hesitate to Get in touch with MaSH to see if there are still options for you.
Sample for Imaging MS
Tissue preparation – fresh frozen preferred, options for fixed/FFPE
Section thickness – ~5-50 microns
Sample plate – MALDI plate, ITO slide, glass slide (MS instrument dependent)
MALDI matrix – choice dictates which biomolecules will be detected
Matrix application – Vacuum sublimation and robotic sprayer available
Sample for Elemental MS
Solution ICP-MS (liquid samples)
Solution ICP-MS analysis is mostly suitable for all* elements except for the following: H, He, C, N, O, F, Ne, Ar, Kr, Xe, Rn. It is not ideal for Cl, Br, I.
Laser ablation ICP-MS (solid samples)
ASI Resolution 193nm laser system (installed May 2019)
Laser Ablation ICP-MS is able to determine the same elemental suite plus Carbon.
Detection Limit: parts per trillion – parts per million (dependent on analysis mode and sample matrix)
Small molecules analysis on LC-MS
This instrument is not suitable for the analysis of inorganic materials. Therefore a desalting step is advised prior to sample submission.
De novo assay setup is time-consuming and hence expensive, so should only be undertaken where large numbers of the sample need analysing.
Volatiles analysis on GC-MS
Sequential derivatization method will be applied to the dried sample to mask most of the active hydrogens (e.g. OH, SH, and NH) of the molecule. This procedure enhances volatility of the sample which leads to improvement of the chromatography resolution and broadens the range of detectable molecules.
For Liquid samples like juice and wine. Do the pilot run with different volumes depending on the sugar content.